Introduction
Les équations d'état sont des relations très importantes en Génie des Procédés, puisqu'elles permettent d'identifier les états physiques de la matière et de calculer les propriétés thermodynamiques de ces systèmes dans leurs états d'équilibres.
Objectif du chapitre 4 :
établir les relations qui existent entre les potentiels d'interaction microscopiques au sein d'un système et les différents termes des équations d'état.
En particulier, nous allons voir comment dériver les équations d'état à partir de la fonction de partition du système, qui incorpore les contributions de l'énergie cinétique et de l'énergie potentielle ; cette dernière dépendant des potentiels d'interaction microscopique.
utiliser les principes et relations de la thermodynamique statistique pour déduire les grandeurs thermodynamiques (U=E, A, G, H, S) et P la pression qui est habituellement employée pour exprimer une équation d'état.
appliquer ces relations pour les gaz parfaits, mono, di et polyatomiques.
survoler des équations d'état usuelles en génie des procédés, cubiques généralisées (SRK, PR, ...) et de type SAFT afin d'illustrer comment les différents termes .
Dans ce chapitre nous choisissons arbitrairement de travailler avec l'ensemble statistique canonique NVT dont la fonction de partition est notée ZN. Mais les mêmes résultats sont obtenus dans tous les autres ensembles statistiques !
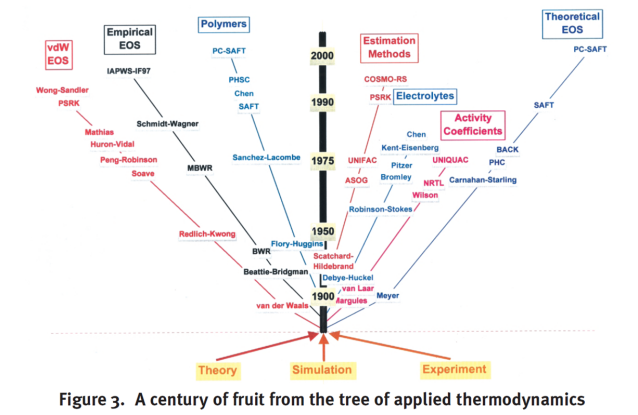
Dans le chapitre introductif, nous avons vu dans l'arbre schématisant un siècle de développement de la thermodynamique appliquée qu'il existe de très nombreuses équations d'état.
Dans ce chapitre, nous nous limiterons aux équations d'états simples équation d'état du gaz parfait, équation de van der Waals
A la fin du chapitre, un petit résumé présente les autres équations d'état cubiques dérivées de celle de van der Waals et également les équations théoriques des fluides associées (SAFT Softly Associated Fluid Theory)